Abstract
Informational genes such as those encoding rRNAs are related to transcription and translation, and are thus considered to be rarely subject to lateral gene transfer (LGT) between different organisms, compared to operational genes having metabolic functions. However, several lines of evidence have suggested or confirmed the occurrence of LGT of DNA segments encoding evolutionarily variable regions of rRNA genes between different organisms. In the present paper, we show, for the first time to our knowledge, that variable regions of the 18S rRNA gene are segmentally replaced by multiple copies of different sequences in a single strain of the green microalga Prototheca wickerhamii, resulting in at least 17 genotypes, nine of which were actually transcribed. Recombination between different 18S rRNA genes occurred in seven out of eight variable regions (V1–V5 and V7–V9) of eukaryotic small subunit (SSU) rRNAs. While no recombination was observed in V1, one to three different recombination loci were demonstrated for the other regions. Such segmental replacement was also implicated for helix H37, which is defined as V6 of prokaryotic SSU rRNAs. Our observations provide direct evidence for redundant recombination of an informational gene, which encodes a component of mature ribosomes, in a single strain of one organism.
- gDNA, genomic DNA
- LGT, lateral gene transfer
- LSU, large subunit
- MP, maximum-parsimony
- mPCR, multiplex PCR
- PHT, partition homogeneity test
- SH, Shimodaira–Hasegawa
- SSU, small subunit
-
↵†Present address: Laboratory of Aquatic Molecular Biology and Biotechnology, Graduate School of Agricultural and Life Sciences, The University of Tokyo, Bunkyo, Tokyo 113-8657, Japan.
-
The GenBank/EMBL/DDBJ accession numbers for the SSU rRNA sequences of Prototheca wickerhamii identified in this work are AB251576–AB251599.
-
Two supplementary figures showing the primers used for mPCR and RT-PCR, and secondary structure models for mosaic 18S rRNA helices, are available with the online version of this paper.
Edited by: D. J. Scanlan
INTRODUCTION
Since the ribosome is a complex piece of machinery for protein synthesis, the sequence homogeneity of the multiple copies of rRNA genes in a single organism (hundreds or thousands in eukaryotes) is essential for correct folding of rRNA molecules and for their precise interaction with other ribosomal components (Jain et al., 1999; Long & Dawid, 1980). It has been suggested that concerted evolution by gene conversion maintains the sequence homogeneity of the multiple copies (Hillis et al., 1991). However, there are several examples of intragenomic nucleotide variations between two distinct types of the same rDNA species (Carranza et al., 1996; Mylvaganam & Dennis, 1992), some of which displayed growth-stage-specific transcription (Gunderson et al., 1987; Mashkova et al., 1981). Furthermore, intragenomic, multiple 18S rRNA gene sequence variations have been detected in primitive fish as well as in diatoms, although no evidence for recombination among these genes has been obtained (Alverson & Kolnick, 2005; Krieger & Fuerst, 2002, 2004; Krieger et al., 2006).
More recently, it has been suggested that small subunit (SSU) rRNA genes underlie the introgression and recombination of gene fragments between closely related but different organisms (Boucher et al., 2004; Dewhirst et al., 2005; Wang & Zhang, 2000). Miller et al. (2005) demonstrated that a chlorophyll d-producing cyanobacterium acquired a fragment of the SSU rRNA gene encoding a structurally conserved hairpin from a proteobacterial donor. This analysis was performed by computational statistical comparison of two likelihood models to account for the identical sequences in variable region 1 (V1) of the SSU rRNA genes of the cyanobacterium and certain proteobacteria: one for lateral gene transfer (LGT) and the other for convergent evolution. Recombination of SSU rRNA gene fragments by LGT would create mosaic rRNAs and affect the evolutionary history recorded in the sequence.
Wang et al. (1997) compared nucleotide sequences of the internal transcribed spacer (ITS) in three rRNA gene operons isolated from a single strain of the actinomycete Thermobispora bispora and found a trace of one segmental replacement consistent with LGT between operons. For eukaryotes, Buckler et al. (1997) also found intragenomic recombination between divergent ribosomal paralogues in ITS regions of land plants. Although the ITS functions in the assembly of ribosomes (King et al., 1986), it is excised from a precursor rRNA and does not serve as a member of the mature ribosomal components that are required to have precise interactions with each other to drive the protein synthesis machinery. In spite of such studies, there has, to our knowledge, been no report of evidence for intragenomic segmental replacement among rRNA genes that encode mature ribosomal components (18S, 26S, 5.8S and 5S rRNA genes in eukaryotes) in a single organism. The present study provides what we believe to be the first example of large-scale recombination of 18S rRNA gene segments between their multiple copies within a single organism, a strain of the non-photosynthetic, unicellular green microalga Prototheca wickerhamii.
METHODS
Algal cultures for isolation of total RNA and genomic DNA.
An axenic culture of Prototheca wickerhamii ATCC 16529 was purchased from the American Type Culture Collection. The lyophilized algal culture was recovered on Sabouraud Dextrose Agar (SDA) plates (Difco). An initial culture was subjected to four successive rounds of single-colony isolation with subsequent subculturing on SDA plates. Single colonies from these cultures were examined for the absence of visible contaminants using a phase-contrast microscope (IX70, Olympus).
18S cDNA and genomic DNA (gDNA) libraries.
Both libraries were constructed as previously described (Ueno et al., 2005). The cDNA pools were derived from total RNA extracted from an early stationary phase culture of the alga grown in Sabouraud liquid medium (Difco). To remove precursor rRNAs, 18S rRNAs were purified from total RNA by gel excision without UV shadowing. The absence of group I introns in 18S rRNA has been shown experimentally (Ueno et al., 2005).
PCR, RT-PCR and mPCR.
AccuPrimeTaq DNA Polymerase High Fidelity (Invitrogen) was used in all types of PCR. The use of the high-fidelity DNA polymerase not only excludes PCR errors but also avoids artificial recombination of the homologous gene fragments (Bradley & Hillis, 1997). PCR was performed in the presence of AccuPrime protein to avoid non-specific annealing between primers and template DNA.
Confirmation of the absence of spliceosomal introns in helix H37.
Helix H37 of the studied strain includes relatively large DNA fragments, of 48–56 bp, which are inserted as a single block in its 18S rRNA genes. These insertions interrupt an evolutionarily conserved region in an otherwise strictly length-conserved part, namely, right at the beginning of the loop in helix H37. Although these insertions are too small, and moreover lack the structural similarities to be considered as group I introns (Dávila-Aponte et al., 1991; Jackson et al., 2002), they may represent spliceosomal introns (Cubero et al., 2000). In order to examine the nature of these small insertions in H37, RT-PCR analysis was performed. Preparation and reverse transcription of 18S rRNAs were performed as previously described (Ueno et al., 2005). A 270–280 bp region containing helix H37 was amplified by PCR with a set of primers shown in Supplementary Fig. S1. The size of each amplicon from cDNA was compared with that of the corresponding gDNA by electrophoresis.
Multiplex PCR (mPCR) analysis.
Four fragments of 18S rRNA genes were simultaneously amplified from different clones of the full-length 18S cDNA and gDNA libraries using two distinct primer mixtures, R and G. Both contained four forward primers that were designed from two distinct sequences (Fig. 1a⇓, red or green boxes) for helices E23-3, H29, H37 and H45/E45-1, together with a shared conserved reverse primer. The primer sequences are shown in Supplementary Fig. S1. See Fig. 1⇓ legend for further details.
Detection of 15 genotypes for 18S rRNA genes within a single algal strain, P. wickerhamii ATCC 16529, based on mPCR. (a, b) Strategy for detecting combination patterns of two distinct sequences in each of four different helices by simultaneous amplification of four DNA fragments from individual clones of 18S rRNA genes using primer cocktails R and G. All forward PCR primers were designed to reflect mosaic gene sequences identified from comparative analyses of three 18S gDNA clones, A–C. Red and green boxes indicate that at least two distinct nucleotide sequences are present for individual helices of the 18S gDNAs. Two different primer mixtures (cocktails R and G) contained four forward primers that were based on two distinct sequences for each of the individual helices E23-3, H29, H37 and H45/E45-1 (red and green arrows) and a shared reverse primer (blue arrows) based on a conserved region within clones A–C (blue boxes). (b) Example of an amplified DNA fragment pattern of mosaic 18S rRNA genes from a single clone obtained by mPCR. (c) Genotypes for 18S rRNA genes were identified based on the presence or absence of amplicons for each of the 18S cDNA or gDNA clones by mPCRs with primer cocktail R (upper) and G (lower). A total of 200 clones (100 each from cDNA and gDNA libraries) were subjected to mPCR-based genotyping. The fragment patterns were reproducible in three mPCR runs for each clone. Different genotypes (1–15) are indicated at the top, and the target helices are shown on the left. M, molecular mass markers. Schematic genotypes are represented below the gel images by the combination of primer types (R and G) that enabled amplification of gene fragments. Similarly, combinations of primer types for clones A and C obtained in our initial analysis are shown on the right, whereas clone B was classified as genotype 10. Identical combination patterns do not represent completely identical sequences. Symbols: red filled circles, fragments amplified by cocktail R; green filled circles, fragments amplified by cocktail G. When both R and G primers did not work due to possible base mismatches between primers and primer annealing sites in the clones, the target regions are indicated by empty circles. The circles in parentheses indicate the presence of only one or two nucleotide mismatches between template DNAs and primers at or near the 5′ end of their annealing sites. In all mPCRs, at least 15 bases at the 3′ end of the primers correctly annealed with the template. Primer sequences are shown in Supplementary Fig. S1. Helices E23-3 of clone g8 and H29 of clone g10 were not detected with cocktail G, although there was no nucleotide mismatch between template DNA and primer (indicated as green parenthesis enclosing an open circle for g8 and as G for g10). The asterisk indicates that the gDNA clone representing genotype 3 was not sequenced due to a loss of the plasmid insert during bacterial culture. Numbers below the names of the representative clones indicate the relative proportion of clones (%) assigned to each genotype.
Phylogenetic analyses and secondary structure prediction.
Full-length 18S rRNA gene sequences of 24 different clones of P. wickerhamii ATCC 16529 were determined. Of these, three clones, A, B and C, were arbitrarily selected from several gDNA clones prepared in our preliminary analysis. After 1 year of algal subculture, we constructed 18S cDNA and gDNA libraries, which comprised a large number of clones. The remaining 21 clones were selected from both libraries to represent ladder patterns (genotypes) as revealed by mPCR (Fig. 1c⇑). The SSU rRNA gene sequences determined in this work have been deposited in GenBank with accession numbers AB251576–AB251599.
Full-length sequences of the 18S rRNA gene were aligned according to their 18S rRNA secondary structure models (Wuyts et al., 2000). Sequence identity within a given segment was checked by creating consensus sequences, and clones with identical sequences within one of the 10 segments examined were grouped together (Fig. 3⇓). Phylogenies based on the segment-specific sequence datasets were inferred by a heuristic search with the maximum-parsimony (MP) analysis. Nodal support was estimated by bootstrap analyses (1000 replicates) according to Felsenstein (1985).
Bayesian analysis was conducted using MrBayes 3.0b4 (Huelsenbeck & Ronquist, 2001) using the general time reversible plus gamma distribution (GTR+Γ) model. The MrBayes program estimates the relative base substitution rates and the value of the shape parameter (α) for the gamma distribution. Four simultaneous Monte Carlo chains were run for 1 000 000 generations. Trees were sampled every 100 generations, yielding 10 000 trees. A consensus tree was created with a burn-in of 200 000 generations equalling 2000 sampled trees; that is, the first 2000 trees were ignored when the consensus tree was created. The trebouxiophytes Auxenochlorella protothecoides SAG 211-7a (X56101) and P. wickerhamii SAG 263-11 (X74003) were used as outgroups in both MP and Bayesian analyses.
A partition homogeneity test [PHT; incongruence length difference (ILD) test; Farris et al. 1994] and Shimodaira–Hasegawa (SH) test (Shimodaira & Hasegawa, 1999) were carried out in paup* 4.0b10 (Swofford, 2002) to test for topological incongruence between the segment-specific phylogenies to infer recombination between different 18S rRNA gene copies.
All of the segment-specific datasets were combined and used for the PHT. The test consisted of 1000 random partitions with heuristic searches using simple addition of sequences and a branch-swapping algorithm (tree-bisection-reconnection). The SH tests were performed using the resampling estimated log-likelihood (RELL) optimization and 1000 bootstrap replicates to determine whether there was a significant difference between tree topologies obtained from individual segment-specific datasets and to compare the MP results with alternative hypotheses based on the enforcement of the following topological constraints: (1) monophyly of the gDNA clones whose gene fragments in H45/E45-1/H46 were amplified by primer cocktail R in mPCR analysis, and (2) the unconstrained tree with the best −ln likelihood score inferred from the segment-specific alignment for H45/E45-1/H46. We employed these constraints to reflect our observations of mPCR results (see Results and Discussion). The SH tests were employed for each of the segment-specific datasets.
P. wickerhamii SAG 263-11 was used as a reference strain for the presentation of predicted secondary structure of 18S rRNA for ATCC 16529 (Fig. 2a⇓). P. wickerhamii was defined on the basis of its ability to utilize trehalose as a sole source of carbon for growth (Pore, 1998). Although a sister relationship between ATCC 16529 and SAG 263-11 was not resolved in the phylogenies based on 18S and 26S rRNA gene sequences, both strains were placed within one clade containing all species of the non-photosynthetic genus Prototheca and a photosynthetic analogous strain A. protothecoides SAG 211-7a (Ueno et al., 2005). P. wickerhamii SAG 263-11 has a single 18S rRNA gene sequence (GenBank accession number X74003).
Secondary structure models for 18S rRNAs of P. wickerhamii ATCC 16529. (a) Clone A compared to P. wickerhamii SAG 263-11. The recombination hot spots identified by LDhat analyses are indicated by arrows. (b) Clone B compared to clone A. (c) Clone C compared to clone A. (d) Clone C compared to clone B. The nucleotides in black, blue, and red boxes indicate base substitutions, insertions and deletions, respectively. The models drawn were based on a multi-sequence alignment of green algal 18S rRNA genes.
Estimation of recombination rate across 18S rRNA genes.
A likelihood-permutation test (McVean et al., 2002) was performed to determine if the observed recombination rate was significantly different from zero. Estimation of variable recombination rates was performed based on an infinite-allele model (McVean et al., 2004), using a penalized likelihood within a Bayesian reversible-jump Markov chain Monte Carlo scheme. The parameters used were as follows: block penalties of 0, 5 and 20, a starting value of 93 for population recombination rate for the entire sequence (2Ner), and 10 000 000 iterations. We sampled every 2000 iterations and ignored the first third of the iterations. Neither different starting values for 2Ner nor longer runs had a significant effect on the estimates. The permutation test and estimation of variable recombination rates were performed using the LDhat package (version 2.0; McVean et al., 2004).
RESULTS AND DISCUSSION
Mosaic structures of 18S rRNA genes from a single Prototheca strain
Initially, we analysed several genomic clones encoding 18S rRNA from a single strain (ATCC 16529) of P. wickerhamii, yielding three typical clones (A–C) with different segmental sequences in helices E23, H29, H37 and E45-1 of variable regions V4, V5, V6 and V8, respectively (Fig. 1a⇑; see Fig. 2a–d⇑ for the secondary structures). The three clones were relatively large (2062–2069 bp) compared to typical 18S rRNA genes of other green algae (e.g. 1802 bp in P. wickerhamii SAG 263-11; Fig. 2a⇑). Most of the base insertions were found in the evolutionarily variable regions V1–V9 (Wuyts et al., 2000). None of them accounted for group Ι (Dávila-Aponte et al., 1991; Jackson et al., 2002; Ueno et al., 2005) or spliceosomal introns (Cubero et al., 2000), which was confirmed by PCR of first-strand cDNAs synthesized from purified 18S rRNA. Clones A and B had almost identical sequences with respect to helices H29 (V5) and E45-1 (Marin et al., 2003) (V8), with only one nucleotide substitution in each of the two helices (Figs 1a⇑ and 2b⇑), but were markedly different from each other in a large helix E23 (V4: ∼300 bp) and H37 (V6: ∼100 bp). On the other hand, the sequence of helix H37 in clone C was identical with that of clone A (Figs 1a⇑ and 2c⇑), helix E23 of clone C was identical to that of the corresponding region of clone B, except for two nucleotide variations (Figs 1a⇑ and 2d⇑), and helices H29 and E45-1 were different from both clone A and clone B. These mosaic gene sequences could be explained by combination of two distinct segmental sequences for the four helices E23, H29, H37 and E45-1, which are shown as red and green boxes in Fig. 1(a)⇑. Thus, we predicted that the multiple 18S rDNA copies in this strain are composed of either ‘red’ or ‘green’ segments for individual helices. While such segmental replacements were also identified in other helices, these four long helices provided obvious and indisputable mosaic structures.
To examine our prediction, cDNA and gDNA libraries of 18S rRNA were constructed, and the ‘red’ and ‘green’ segmental sequences in the four helices of 100 clones from each of the two libraries were mPCR-amplified with two different primer cocktails, R and G (Fig. 1b⇑). The primer cocktails R and G contained four forward primers that were specific to one of two distinct sequences (red and green boxes) found in helices E23-3, H29, H37 and H45/E45-1 of the three gDNA clones, A, B and C. The first primer cocktail, R, contained only ‘red’ (forward) primers and the second cocktail, G, contained only ‘green’ (forward) primers; both primer sets were combined with a shared reverse primer that was based on an evolutionarily conserved region near the 3′ end of the 18S rRNA gene, as indicated by the blue boxes in Fig. 1(b)⇑. The primer sequences are listed in Supplementary Fig. S1.
Fig. 1(c)⇑ shows mPCR-based genotypes for the 18S rRNA gene obtained with a combination of ‘red’ (R) and ‘green’ (G) primers that enabled amplification of helix E23-3, H29, H37 or H45/E45-1. Both R and G primers failed to amplify at least one out of the four helices for 170 clones (empty circles in Fig. 1c⇑), probably due to unique sequences in the primer regions, which might be different from those originally found in clones A–C. Nevertheless, 15 genotypes (Fig. 1c⇑) were clearly identified, although those corresponding to clones A and C obtained in the initial analysis (Fig. 1a⇑) were absent in the 200 clones examined. The sequence of clone B corresponded to that found in one of the genotypes (genotype 10). Seven genotypes were found in both libraries, whereas six and two genotypes were found only in gDNAs and cDNAs, respectively (Fig. 1c⇑). One representative clone for each genotype was sequenced, whereas two representative clones, one from cDNAs and the other from gDNAs, were sequenced when a single genotype was found in both libraries. cDNA and gDNA clones from a single genotype revealed nucleotide variations distributed over the whole length, with nucleotide identities of 95.2–98.8 % using a secondary-structure-based alignment. A total of 21 clones from the cDNA and gDNA libraries were sequenced (Fig. 1c⇑) and none of them had identical sequences. One clone was not subjected to sequencing due to a loss of insert DNA in the plasmid during bacterial culture. In summary, a total of 24 different sequences were identified when clones A–C obtained in the first analysis were included. More different sequences would be expected if different clones of the same genotype were to be analysed. The overall nucleotide identities among 24 different 18S rRNA gene sequences were markedly low in some cases, ranging from 89.7 to 98.9 % (pairwise comparison using the secondary-structure-based alignment). Five genotypes (1–5) in both cDNA and gDNA libraries were the most predominant, whereas six genotypes (10–15), accounting for 13 % of the gDNA clones, were not found in the cDNA library (Fig. 1c⇑). Some 18S rRNA genotypes with particular segmental replacements in their helices might not be transcribed, although the secondary structures of almost all gDNA clone sequences showed apparent local compensating base substitutions in individual helices, except in one case described below.
Extent of intragenic segmental replacement
Next, we inferred phylogenies of the 24 different 18S rRNA genes based on their partial sequences at the level of recombinant segments to investigate the extent of intragenic recombination. We aligned the 18S rRNA sequences of the 10 most variable segments (three segments from V2 and one segment each for V3–V9), and constructed phylogenetic trees based on the sequence alignment for individual segments. The optimal MP topologies using the SH test are shown in Fig. 3⇓ (V1 sequences showed no nucleotide variation). These segments do not necessarily correspond to the helices in the evolutionarily variable regions. The results for E23 (V4), H29 (V5), H37 (V6) and H45/E45-1/H46 (V8) are shown in Fig. 3(a)⇓. The trees corresponding to the remaining six variable segments are shown in Fig. 3(b)⇓. The topologies of the segment-specific phylogenies shown in Fig. 3(a)⇓ are consistent with the classification of the clones as revealed by mPCR with R and G primers (see Fig. 1⇑). Several gDNA clones of genotypes 10–15 formed one clade for H17, H29, H37, H43, H45/E45-1/H46 and H49 (Fig. 3⇓, lines in magenta); each clade (except for H17) was supported by values greater than 50 % for both bootstrap proportions using the MP method and Bayesian posterior probability. However, these gDNA clones did not form one clade for E23 (Fig. 3a⇓) and for three segments in V2 (Fig. 3b⇓). These observations prompted us to statistically compare alternative hypotheses by the SH test, focusing on the monophyletic lineage of gDNA clones from genotypes 10–15 in which none of the cDNA clones was included. We enforced constraints that reflected the topology of the unconstrained tree with the best −ln likelihood score based on the dataset for segment H45/E45-1/H46, because a lineage of the clones of genotypes 10–15 for this segment received higher bootstrap/posterior probability values than those for the other segments (Fig. 3⇓). Although enforcing topological constraints resulted in a significant increase in the −ln likelihood scores in all segments, the alternative topologies that force the monophyly of the aforementioned gDNA clones were not significantly worse than the best trees (Fig. 3⇓, 0.267<P<1.000). Therefore, monophyly of the clones of genotypes 10–15 was not rejected.
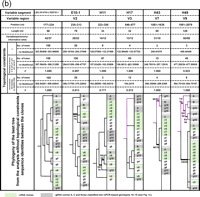
MP trees inferred from the sequence alignment for variable segments in helices E23, H29, H37 and H45/E45-1/H46 (a), and in L9/L10-H10-L10/E10-1, E10-1, H11, H17, H43 and H49 (b) of 18S rRNA genes from P. wickerhamii ATCC 16529. The trees represent optimal topologies derived from unconstrained analyses in the SH tests. Sequences of 24 different clones were used, of which 21 were determined in the mPCR experiments (Fig. 1c) and three (clones A–C) in the initial analyses. Trees were rooted using the sequences of the trebouxiophytes A. protothecoides SAG 211-7a (X56101) and P. wickerhamii SAG 263-11 (X74003). Groups of clones between lines had identical sequences within the respective segments. Bootstrap proportion values (%) using the MP method (first number) and Bayesian posterior probability support (second number) are shown at internal nodes on thick bold branches, only when both values are greater than 50 %. Partial 18S rRNA gene sequences that corresponded to individual segments were aligned by considering the secondary structure. Nucleotide numbering refers to the secondary structure-based alignment including alignment gaps of full-length sequences for 24 different 18S rRNA genes of ATCC 16529, plus those for SAG 211-7a and SAG 263-11. Numbers of variable sites and those of informative sites for MP analysis, as well as numbers of equally parsimonious trees compared, −ln likelihood scores, and P values calculated in the SH tests, are shown above the phylogenies. cDNA clones are boxed in pale green. The lines in magenta represent monophyletic lineages of gDNA clones that do not include cDNAs (in segments for helices H17, H29, H37, H43, H45/E45-1/H46 and H49).
In contrast, a PHT among the 10 variable segments was significant (P=0.001), indicating incongruence. Similarly, a significance test for recombination based on a permutation of the likelihood (McVean et al., 2002) gave significant correlation between the r2 measure of linkage disequilibrium (LD) and physical distance (r2: −0.31017, P<0.000), as well as correlation between the |D′| measure of LD and physical distance (|D′|: −0.24332, P<0.000), rejecting the hypothesis of no recombination. Fig. 4⇓ shows recombination intensity estimated across 24 different 18S rRNA genes. The LDhat analyses with a block penalty of 0 estimated elevated recombination activities in most of the 10 variable segments shown in Fig. 3⇑. The recombination hot spots are also indicated by arrows in the secondary structure model for clone A (Fig. 2a⇑), showing that they are located in or near the elongate stem–loop structures in the variable segments. Although peaks for elevated levels of recombination were not inferred for H10, H17 and H43, they were located in the regions where significant changes in the recombination rates were observed. Larger penalties of 5 and 20 were inadequate for estimation of increased recombination activities in H11 and H37. In addition to these helices, our exhaustive search for recombination without block penalties found two more hot spots with increased recombination intensities in E23 and one more in H49. Large block penalties identify positions where strong evidence is found for changes in recombination rate, but would ignore positions where slight changes in the rate are found in the analyses without penalty.
Estimated recombination rate across 18S rRNA genes of P. wickerhamii ATCC 16529 for block penalties of 0 (dark blue), 5 (pink) and 20 (green). The population recombination rate 2Ner was estimated across the full-length 18S rRNA genes using LDhat (McVean et al., 2002). Blue arrows indicate elevated recombination activities estimated for the positions which belong to the variable segments shown in Fig. 3. These segments are indicated by the thick horizontal lines below the x axis.
In summary, all 24 clones analysed showed discordant phylogenies for the variable segments, suggesting that these segments were replaced among different 18S rRNA genes, with the exception of clones from genotypes 10–15, for which clear evidence of recombination was not obtained with the SH test. The replaced segments were found to be mostly located in the inserted regions compared with strain SAG 263-11 of the same species (Fig. 2a⇑).
Segmental replacement of DNA sequences among different 18S rRNA genes was observed even in the short fragments of helices H29, H37, and E45-1 (Fig. 5⇓). The flow in such short fragment replacement is illustrated in Supplementary Fig. S2 available with the online version of this paper. Helix H29 contained two mosaic sequences with different short fragments derived through inter-helical segmental replacement, each of which behaved as a single cassette at an identical location (Figs 3a⇑ and 5⇓, and Supplementary Fig. S2a, b). Thus, helix H29 (V5) has three different recombination loci, including that for segmental replacement of the whole helix region. The mosaic sequences in H29 and E45-1 can be folded into a putative secondary structure, whereas no reasonable structure is obvious for H37 of clone g12 (Figs 3a⇑ and 5⇓, and Supplementary Fig. S2c). In contrast to the results from the SH test, however, short fragments of the gDNA clones of genotypes 10–15 were found to participate in recombination, as far as helices H29, H37 and E45-1 are concerned (Fig. 5⇓, Supplementary Fig. S2).
Formation of short mosaic fragments in helices H29, H37 and E45-1. Names of chimeric clones are underlined. The nucleotide sequences of the donor clones are either in lower-case blue or upper-case red type. In chimeras, donor sequences are identified by blue or red type. See Fig. 3 for phylogenetic position of individual clones. See Supplementary Fig. S2 for the secondary structure models of helices H29 and H37 displaying short mosaic fragments.
Two distinct causative mechanisms of 18S rRNA gene sequence variation in a single Prototheca strain
The sequence alignment for 24 different 18S rRNA gene copies in P. wickerhamii ATCC 16529 revealed 36 base-substitution sites in regions that are conserved in 90–100 % of all green algae and land plants studied so far. All but one of the 36 base substitutions were found only in one gene copy out of 24; they were not concentrated in particular clones. These substitutions therefore did not originate from segmental replacement. They accounted for 34 transitions and two transversions, while in evolutionarily variable regions, especially in the segmentally replaced ones, 111 transitions and 130 transversions were found. This is in agreement with the results of a previous comparative analysis of SSU rRNA genes in Streptomyces strains in which two substitution patterns were observed (Ueda et al., 1999). One included single random base exchanges, which were mainly transitions, and the other one contained fragments consisting of five or more base exchanges, which mainly involved transversions, suggestive of LGT. Artificial recombination during PCR (Bradley & Hillis, 1997) could be excluded for the present segmental replacements based on several lines of evidence. For example, (1) segmental replacement was limited to variable regions, (2) the replaced regions have partially divergent sequences, and (3) the overall secondary structure was maintained. Therefore, intragenomic recombination of rRNA gene segments is considered to function in the formation of mosaic 18S rRNA genes in P. wickerhamii. However, recombination of homologous genes between alternative alleles is excluded, as P. wickerhamii is haploid and reproduces asexually by autosporulation.
Conclusion and perspectives
We have provided direct evidence for large-scale recombination of an informational gene in a single strain of one haploid organism. This gene encodes one component of mature ribosomes, 18S rRNA, that has been thought to be recalcitrant to LGT. In P. wickerhamii ATCC 16529, 18S rRNA genes are shaped as a complex mosaic of divergent gene fragments which are located in most of the evolutionarily variable parts of this gene. Large helices as well as short intrahelical gene fragments are involved in the mosaic structure. Compared to the large-scale intragenomic segmental replacement of 18S rRNA genes observed here (Fig. 4⇑), loci described to date for recombination of SSU rRNA gene fragments between different organisms have been restricted to (1) short gene segments, (2) only one conserved hairpin, or (3) two or three regions that were likely to correspond to conserved stems. In the first case, a comparative sequence analysis of the SSU rRNA genes of various actinomycetes has suggested recombination of short gene segments between species (Wang & Zhang, 2000). These segments correspond to only three to five base pairs in the stems of certain hairpins of the RNA product. Furthermore, statistical comparison of alternative likelihood models (representing LGT or convergent evolution) was not performed. Therefore, it is not clear whether or not LGT is responsible for the occurrence of identical base pairs in those hairpins of SSU rRNA in different actinomycetes. In the second case, two strains of a chlorophyll d-producing cyanobacterium have been found to have gained a single hairpin helix in variable region V1 of the SSU rRNA from a β-proteobacterium (Miller et al., 2005). In the third case, comparative sequence analysis of six rRNA operons of the actinomycete Thermomonospora chromagena has revealed that the SSU rRNA gene of one operon (rrnB) contains two or three regions that were likely derived from a different species, Thermobispora bispora, whereas sequences of the corresponding genes from the remaining five operons in T. chromagena (rrnA, rrnC, rrnD, rrnE and rrnF) are almost identical with each other and lack evidence for interspecific recombination (Yap et al., 1999; Gogarten et al., 2002).
It is unclear how recombinant 18S rRNA genes are created in the genome of the specific strain (ATCC 16529) of P. wickerhamii studied here. As P. wickerhamii is haploid, crossover of 18S rRNA genes by homologous recombination is not to be expected. The origin of different types of SSU rRNA gene in a single genome has been explained by either (1) divergent evolution following gene duplication (Mylvaganam & Dennis, 1992; Baliga et al., 2004) or (2) interspecific LGT (Wang & Zhang, 2000; Gogarten et al., 2002; Miller et al., 2005). Even if one of these scenarios did apply to strain ATCC 16529, it ought to be followed by further recombination of divergent gene fragments between intragenomic operons. Specifically, recombination is obviously necessary to form mosaic genes in the first scenario, but also in the second to explain the observed highly segmental nature of 18S rRNA genes, as loci for recombination via LGT between organisms are restricted in SSU rRNA genes (Wang & Zhang, 2000; Gogarten et al., 2002; Miller et al., 2005). In any case, it is plausible that a mobile genetic element-like mechanism (which works in a single genome) might play a role in the formation of the complex mosaic structure observed here.
It was not possible to determine whether the redundant recombination events have a positive effect on ribosome function in the present strain. Most of the 18S rRNA variants retained a reasonable secondary structure. Nevertheless, six genotypes of the gDNA clones were not found in the cDNA library according to the mPCR-based genotyping of the 18S rRNA genes. Therefore, the potentially crucial biological function that the organism pursues via redundant segmental replacement of 18S rRNA genes has yet to be identified. Fixation and long-term maintenance of a single foreign gene fragment in an SSU rRNA gene was thought to have been favoured by natural selection (Miller et al., 2005), but this does not explain further recombination of 18S rRNA genes in a single genome. Sweeney et al. (1996) inserted DNA fragments, which were complementary to the coding strands of protein-encoding genes, in the variable region of the large subunit (LSU) rRNA gene of the protist Tetrahymena thermophila. The transcribed rRNA, which functioned as an antisense RNA, eliminated target gene expression without impairing the ribosome function itself. Based on this result, those authors proposed the use of rRNA as a vehicle for antisense RNAs. In ATCC 16529, most helices in variable regions of the SSU rRNA (Fig. 2a⇑) were extended compared to a different strain of the same species, SAG 263-11, and the extended regions were segmentally replaced. Although the aim of Sweeney et al. (1996), to insert DNA fragments into variable regions of the rRNA gene, was of a different nature, such a mechanism could be advantageous to maintain precise interactions between 18S rRNA and other ribosome components, if at least one of them is variable with respect to its nucleotide or amino acid sequence.
Much effort is required to investigate the role of variable regions in rRNA genes by using in vivo modification in a model organism (Sweeney et al., 1994; Jeeninga et al., 1997). Such studies have shown that shortening, extension and base changes of variable regions in LSU rRNA may have detrimental or lethal effects on the viability of the organism. In contrast, other studies have described successful construction of functional ribosomes whose components were put together from distantly related species (Nomura et al., 1968; Asai et al., 1999). Therefore, the contribution of variable rRNA regions to proper ribosome function varies between organisms. Considering these observations, P. wickerhamii ATCC 16529 has a significant potential for investigations of its intact, purified ribosomes without any modification of 18S rRNA genes in vitro.
Based on nuclear SSU rRNA gene phylogenies, the non-photosynthetic green algae Prototheca and Helicosporidium, and their photosynthetic analogue A. protothecoides, form a monophyletic group (Ueno et al., 2005). Within this group, poor statistical support for the branching order of the deeper lineages results in uncertain placement of different species and strains, including P. wickerhamii ATCC 16529. As most of these strains form unusually long branches in the tree, this could have caused plesiomorphic long branch attraction effects (Felsenstein, 1988; Ueno et al., 2005). Recurrent recombination of divergent 18S rRNA gene fragments in P. wickerhamii (and possibly in its ancestral organisms) provides one plausible explanation for the rapid rate of evolutionary change in their 18S rRNA gene sequences, leading to ambiguous placements in phylogenetic trees. If the present organism were to discontinue redundant recombination leaving an 18S rRNA gene with a single sequence in its genome, we would merely recognize this as a fast-clock evolving gene without knowing the underlying mechanism. In fact, we are not aware of any other strains of Prototheca species (Prototheca moriformis, Prototheca stagnora, Prototheca ulmea and Prototheca zopfii) with different 18S rRNA genes, although all of them form long branches in phylogenetic trees based on the sequences of this gene (Ueno et al., 2005).
Acknowledgments
We would like to thank Dr Jorge Fernandes (University of St Andrews) for his helpful comments.